|
|
|||||||||||||||||||||
|
|
||||||||||||||||||||||
| ��ǰλ�� ->�� 4��˫ԭ�ӷ��� -> 4.4���ۼ����ۺ�����ӽṹ
-> 4.4.2 ����ӵļۼ����� |

| ��������ӵļۼ����� ��ʮ���Heitler-London��H2�ı�ִ����Ǽۼ����۵Ŀ����Թ�����H2����������ԭ�Ӻ�A��B���������ӣ���ͼ4-5 ��ʾ����������Զ��ʱ����ϵ�Ļ�̬����������ԭ�ӡ����ǿ��Լٶ�����e1�ͺ�A���ϣ�����e2�ͺ�B���ϣ�����ԭ�Ӽ����������Ǻ˼��rAB�ĺ�������rAB������Сʱ����Ѹ�ٵر�Ϊǿ�ҵ��ų�����(�μ�ͼ4-5�е�����)
ͼ4-3 H2+����������ͼ �Ӽ��㿴��������ԭ�Ӳ����ϳ��ȶ��ķ��ӡ������������Ǻ�������һ�ֽṹ��������e2�ͺ�A�Ľ�ϡ�����e1�ͺ�B�Ľ�ϣ�������ٶ��Ľṹ����ͬ���ȶ��ԡ�����������ѧԭ�������Dz�����Ϊ����ijһ�ṹ�ɵ��������������ϵ�Ļ�̬������Ҫ�������ֽṹ���������ӳ��һ��ϵ��
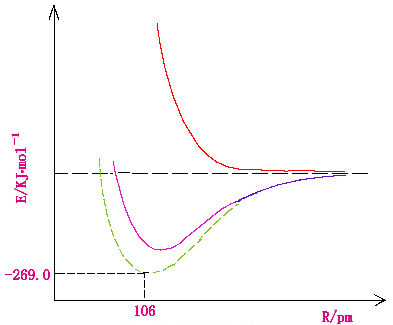
�����ó���������������߾������Եļ�Сֵ(��ͼ�����ʵ��)������
Ϊ�����̽������ ����ӵĹ��ܶ�����ɱ�ʾΪ�������ӵĶ��ܣ�������˵������ܣ�����������ų��ܣ�������ų���֮�͡�
= �ٰ�������ϳ�Haԭ�ӹ��ܶ������Hbԭ�ӹ��ܶ����������������� ���ݱ�ַ�
�����������õļ���ԼΪʵ��ⶨֵ��67%���Ժ����˽�һ���Ľ���̽������������õ��ļ��ܴ���ȷֵ��80%��Pauling �������ϵĴ�������Ϊֻ��������ӵ������������˶��зֱ���ͬ�ĺ˵Ľṹ��δ������������ͬʱ����ij���˵�����������ӽṹIII��IV�� �ṹIII��
(HA:)-H ����ṹ����һ����������H+��һ����������ӽṹ�ĸ�������H�����ڸ�������Ҳ�����˵�����Ե�Ҫ��III��IV�ṹ�ڻ�̬�������Ҳռ��һ�������� ������������������ֽṹ������Լռ�ܼ��ܵ�5%��Pauling
��Ϊ��ʵ����������µ�15%�������ɷ��ӱ�����������ģ���ǰ������к����˸��ӵ�����ã�ֱ��1958��James
�ȶԻ�̬H2���˾�ȷ�����۴�������õķ��Ӽ���Ϊ102.62Kcal/mol����ʵ��ⶨһ�£�ƽ��˼�࣬��Ƶ�ʵ�Ҳ��ʵ�������
|
|||||||||||||||||

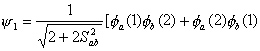
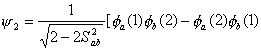
|




