学习向导
练习测试
相关资源
知识点检索
课程介绍
课程目录
当前位置 ->§ 2 原子的结构 ->
2.3 多电子原子结构
-> 2.3.1 中心力场近似和自洽场近似
§2.3 多电子原子结构(The structure of multi-electron atoms)
一、中心力场近似和自洽场近似
(一)、自洽场方法
1.多电子原子的薛定谔方程
氢原子是最简单的原子体系,仅有一个原子核与一个电子。即使这样简单的体系,解薛定谔方程时,还需用特殊函数,如连属勒让特多项式和拉盖尔多项式。对任意一个多电子原子来说,要精确求解薛定谔方程是不可能的,只能寻找某种近似方法。其中比较成功的是中心力场近似。假定原子的质心与原子核心重合,哈密顿算符中,可略去原子核的动能项,并以原子单位表示一些常数
。这样,
化简为:
其中第一项是对原子中所有电子的动能求和,第二项是电子与核间相互作用势能的加和。第三项是电子间的相互排斥势能。近似方法希望能将原子的总哈密顿算符分解成一个个单电子哈密顿算符的加和,薛定谔方程中前两项很容易分解成单电子项,只有第三项难以分解。中心力场近似假设每个电子处在原子核与其它电子组成的平均势场中运动:
此处V(i)是某电子i的单电子势能函数,它以原子核势场、其余(n-1)个电子产生的瞬时场平均值为基础,总波函数ψ写成单电子波函数的乘积:
波函数的平方――几率密度函数
,根据概率论,恰好是单电子几率密度函数
的乘积。因此,用单电子轨道乘积求多电子近似波函数所隐含的物理模型是一种独立电子模型。
体系的总能量近似为各电子能量之和
2.自洽场方法
根据中心力场近似,每个电子都是在一个由核和其它电子产生的平均势场中运动,它的波函数可用
来表示,它们都服从方程
Hartree在1928年首先提出,后经Fock改进的自洽场方法如下:先假设一套试探的单电子波函数
,例如可仿照类氢类子波函数
由径向函数与球谐函数组成,将试探波函数代入Hartree-Fock方程,得到第一次循环后的能量和波函数
,再将波函数
代入Hartree-Fock方程,得到第二次循环后的能量和波函数
,再将波函数
代入Hartree-Fock方程……这样反复计算,直至n次能量与(n-1)次的能量的差值(或波函数形成的密度函数差值)小于某一指定值,则称方程达到自洽。此方法刚开始是解原子的Hartree-Fock方程,以后五,六十年代经Roothann改进后可处理分子体系。Hratree-Fock-Roothann方程:
直至目前,仍是量子化学计算使用的重要方程,自洽场方法随着计算机的普及,变得越来越有效了。

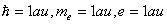 。这样,
。这样, 化简为:
化简为: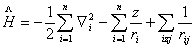
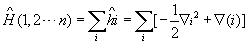
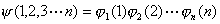
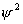 ,根据概率论,恰好是单电子几率密度函数
,根据概率论,恰好是单电子几率密度函数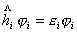 体系的总能量近似为各电子能量之和
体系的总能量近似为各电子能量之和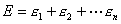
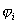 来表示,它们都服从方程
来表示,它们都服从方程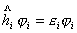
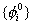 ,例如可仿照类氢类子波函数
,例如可仿照类氢类子波函数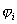 由径向函数与球谐函数组成,将试探波函数代入Hartree-Fock方程,得到第一次循环后的能量和波函数
由径向函数与球谐函数组成,将试探波函数代入Hartree-Fock方程,得到第一次循环后的能量和波函数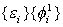 ,再将波函数
,再将波函数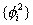 代入Hartree-Fock方程……这样反复计算,直至n次能量与(n-1)次的能量的差值(或波函数形成的密度函数差值)小于某一指定值,则称方程达到自洽。此方法刚开始是解原子的Hartree-Fock方程,以后五,六十年代经Roothann改进后可处理分子体系。Hratree-Fock-Roothann方程:
代入Hartree-Fock方程……这样反复计算,直至n次能量与(n-1)次的能量的差值(或波函数形成的密度函数差值)小于某一指定值,则称方程达到自洽。此方法刚开始是解原子的Hartree-Fock方程,以后五,六十年代经Roothann改进后可处理分子体系。Hratree-Fock-Roothann方程: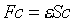 直至目前,仍是量子化学计算使用的重要方程,自洽场方法随着计算机的普及,变得越来越有效了。
直至目前,仍是量子化学计算使用的重要方程,自洽场方法随着计算机的普及,变得越来越有效了。



